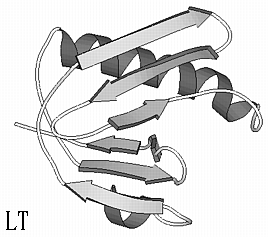
Verotoxin (VT) and Heat-labile Enterotoxin (LT) cause hemolytic uremia and traveller's diarrhea, respectively. From the 3D structures of both toxins it is clear that their B-subunits adopt the same fold, now known as the OB-fold. The RMS deviation of 47 CA is 1.0 Å while only three residues are identical in a structure-based sequence alignment (VT and LT B-subunit sequences are 69 and 101 residues long, respectively). Given the 3D structure of the VT B-subunit, can threading methods identify the fold of LT B?
Sequence alignment based on 3D structures
b b b b b b b b b b b
VT - - - - - - - - - - - - - - - T P D C V T G K V E Y T K Y N D - - -
| | | | | | | |
LT A P Q T I T E L C S E Y R N T Q I Y T I N D K I L S Y T E S M A G K
a a a a a b b b b b b b b b b b b b
b b b b b b b b b b a
VT D D T F T V K V - G D K E L F T N R - - - - - - - - - - - - - W N L
| | | | | | | | | | | | | | | |
LT R E M V I I T F K S G E T F Q V E V P G S Q H I D S Q K K A I E R M
b b b b b b b b b a a a a a a a a a a
a a a a a a a a a a b b b b b b b b b
VT Q S L L L S A Q I T G M T V - T I K T N A C H N G G G F S E V I F R
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
LT K D T L R I T Y L T E T K I D K L C V W N N K T P N S I A A I S M K N
a a a a a a a a a b b b b b b b b b b b b b b b b
The OB-fold is:
10 best structures:
| PDB structure | raw score | Z-score | a.a.aligned | % identites | Fold (class) |
| 1bovAx 69 a.a. | 3062 | 10.40 | 69 | 100 | OB (all beta) |
| 1ltsDx 103 a.a. | 569 | 1.48 | 89 | 16 | OB (all beta) |
| 1mhcBx 99 a.a. | 543 | 1.38 | 81 | 20 | MHC recogn. dom. (alpha + beta) |
| 1ayaAx 101 a.a. | 470 | 1.12 | 79 | 21 | SH2-like(alpha + beta) |
| 1fkj_x 107 a.a. | 434 | 0.99 | 78 | 21 | FKBP-like (alpha + beta) |
| 2mcm_x 112 a.a. | 401 | 0.87 | 82 | 20 | Immunoglob.-like (all beta) |
| 1aac_x 105 a.a. | 386 | 0.82 | 84 | 26 | Cupredoxin-like(all beta) |
| 2fxb_x 81 a.a. | 381 | 0.80 | 75 | 13 | Ferredoxin-like (alpha + beta) |
| 1bdo_x 80 a.a. | 369 | 0.76 | 74 | 16 | ND |
| 1prtDx 110 a.a. | 366 | 0.75 | 84 | 17 | OB (all beta) |
Alignments:
1bovAx 3062 finds itself
bb bbbbb bbbbb bb bbaaaaa
MKKTLLIAASLSFFSASALATPDCVTGKVEYTKYNDDDTFTVKVGDKELFTNRWNLQSLL
****************************************
--------------------TPDCVTGKVEYTKYNDDDTFTVKVGDKELFTNRWNLQSLL
bbbbb bbbbbbbbbbbbbbb aaaaa
aaa bbbbbb bbbbbbb
LSAQITGMTVTIKTNACHNGGGFSEVIFR
*****************************
LSAQITGMTVTIKTNACHNGGGFSEVIFR
aaaaaaabbbbbbb bbbbb
1ltsDx 569 finds Heat-labile enterotoxin
bb bb bbb bbbbb bb
MKKTLLIAAS----LSFFS--ASALATPDCVTGKVE--YTKYNDDDTFTVKVGDKELF-T
.*. * ..|. . * .:. .** * ...|.:.|** *.*.:.: .
APQTITELCSEYRNTQIYTINDKILSYTESMAGKREMVIITFKSGETFQVEVPGSQHIDS
aaaaaaaa bbbbbbb bbbbbbb bbbbbbbb bbbbb
bbaaaaaaaa bbbbbb bbbbbbb
NRWNLQSLLLSAQITGMTVTIKTNAC----HNGGGFSEVIFR-
.. .::.: . .** .* * . . * ..:.:.... .|
QKKAIERMKDTLRITYLTETKIDKLCVWNNKTPNSIAAISMKN
aaaaaaaaaaaaaaaaaaabbbbb bbbbbbb
--- ------------------------------------------------------------
--- TOPITS prediction-based threading
--- ------------------------------------------------------------
--- TOPITS ALIGNMENTS HEADER: ABBREVIATIONS
--- RANK : rank in alignment list, sorted according to z-score
--- EALI : alignment score
--- LALI : length of alignment
--- IDEL : number of residues inserted
--- NDEL : number of insertions
--- ZALI : alignment zcore; note: hits with z>3 more reliable
--- PIDE : percentage of pairwise sequence identity
--- LEN2 : length of aligned protein structure
--- ID2 : PDB identifier of aligned structure
--- NAME2 : name of aligned protein structure
---
--- TOPITS ALIGNMENTS HEADER: ACCURACY
--- : Tested on 80 proteins, TOPITS found the
--- : correct remote homologue in about 30% of
--- : the cases, detection accuracy was higher
--- : for higher z-scores (ZALI):
--- ZALI>0 : 1st hit correct in 33% of cases
--- ZALI>3 : 1st hit correct in 50% of cases
--- ZALI>3.5 : 1st hit correct in 60% of cases
---
--- TOPITS ALIGNMENTS HEADER: SUMMARY
RANK EALI LALI IDEL NDEL ZALI PIDE LEN2 ID2 NAME2
1 89.43 69 0 0 4.25 100 349 1bov_A VEROTOXIN-1 OB fold (all beta>
2 88.48 69 0 0 4.18 100 349 1bov_A VEROTOXIN-1
3 88.43 69 0 0 4.17 100 349 1bov_A VEROTOXIN-1
4 88.28 69 0 0 4.16 100 349 1bov_A VEROTOXIN-1
5 87.87 69 0 0 4.13 100 349 1bov_A VEROTOXIN-1
6 57.28 88 14 5 1.82 19 476 2aaa D alpha-amylase beta dom (all beta)
7 56.65 88 9 2 1.78 16 244 1rva_A restriction endonuclease (alpha/beta)
8 55.65 86 4 2 1.70 16 1275 1tah_A alpha/beta hydrolase (alpha/beta)
9 55.65 86 4 2 1.70 16 1275 1tah_A alpha/beta hydrolase (alpha/beta)
10 55.48 87 15 3 1.69 15 405 1eft EF-Tu C-term Dom. (all beta)
11 55.25 88 18 3 1.67 13 496 1smd L_ ND
12 55.15 80 3 2 1.66 17 1275 1tah_A alpha/beta hydrolase (alpha/beta)
13 54.92 83 14 4 1.65 17 131 1ifc Lipocalins (all beta)
14 54.43 87 5 3 1.61 13 686 1cdg Immunoglobulin-like (all beta)
15 54.10 87 6 4 1.58 22 648 2bbv_A Viral capsid proteins (all beta)
16 53.93 86 4 2 1.57 16 1275 1tah_A alpha/beta hydrolase (alpha/beta)
17 53.77 87 14 2 1.56 17 452 1gcb L_ID Immunoglobulin-like (all beta)
18 53.37 81 13 4 1.53 15 377 1hsb_B Immunoglobulin-like (all beta)
19 53.37 81 13 4 1.53 15 377 1hsb_A Immunoglobulin-like (all beta)
20 53.28 86 6 4 1.52 22 648 2bbv_A Viral capsid proteins (all beta)
The method used for this prediction was: gonnet .
NOTE: This method is merely a sequence-sequence comparison
of your sequence to the sequences of the PDB folds.
Most similar fold: 1bova
VEROTOXIN-1 (B-OLIGOMER, ALSO CALLED SHIGA-LIKE T
RANK Z-SCORE FOLD LENGTHALI %ID
1 49.98 1bova 69 100 2-21 beta ; OB-fold] 2-21-1-1-3-1-1-1
2 4.23 1tiid 77 25 1997 new ; ESCHERICHIA COLI HEAT LABILE ENTEROTOXIN TYPE I]
3 3.72 1dlha_3-81 72 21 4-12 (a+b) ; MHC antigen-recognition domain] 4-12-1-1-2-1-1-1 RC:,
4 3.16 1ifl 52 15 1-71 alpha ; Oligomers of long helices] 1-71-3-1-1-4-1
5 2.96 1drs 51 12 7-5 small ; Snake toxin-like] 7-5-1-2-1-1-1
6 2.89 1erg 64 16 7-5 small ; Snake toxin-like] 7-5-1-3-1-1-1
7 2.84 3hhrb_131-234 86 19 2-1 beta ; Immunoglobulin-like beta-sandwich] 2-1-2-1-5-1-1-2
8 2.80 1mjc 63 24 2-21 beta ; OB-fold] 2-21-5-1-1-1-1
---> Major cold shock protein from Escherichia coli (n=5, S=8)
9 2.55 2cyr 72 26 7-27 small ; Cytochrome c3] 7-27-1-1-1-1-2
10 2.42 1frta_179-269 64 23 2-1 beta ; Immunoglobulin-like beta-sandwich] 2-1-1-2-3-1-2-1
LEGEND:
COL. 1: RANK. The ranks are obtained by sorting the fold library,
by Z-SCORES, in decreasing order. Only the 10
structures that are most compatible to your sequence
are shown.
COL. 2: Z-SCORE. The z-scores are computed using
the distribution of raw scores (not shown) of all folds.
COL. 3: FOLD. Protein Data Bank codes for the coordinates of the 3D
structures.
COL. 4: LENGTHALI. The number of residues from your sequence that were
aligned to the fold.
COL. 5: % ID. Percentage of identical residues in the alignment.
RELIABILITY OF THIS PREDICTION:
With this method the confidence threshold
is a z-score of 4.4 +- 1.0.
![]() 5. Comparative Modeling: From Sequence to 3D model
5. Comparative Modeling: From Sequence to 3D model