| RESEARCH |
| "Protein structure-based drug design is rapidly
gaining momentum. The new opportunities, developments and results in this
field are almost unbelievable ..."
Christophe L.M.J. Verlinde & Wim G.J.
Hol (1994). Structure 2, 577-587.
|
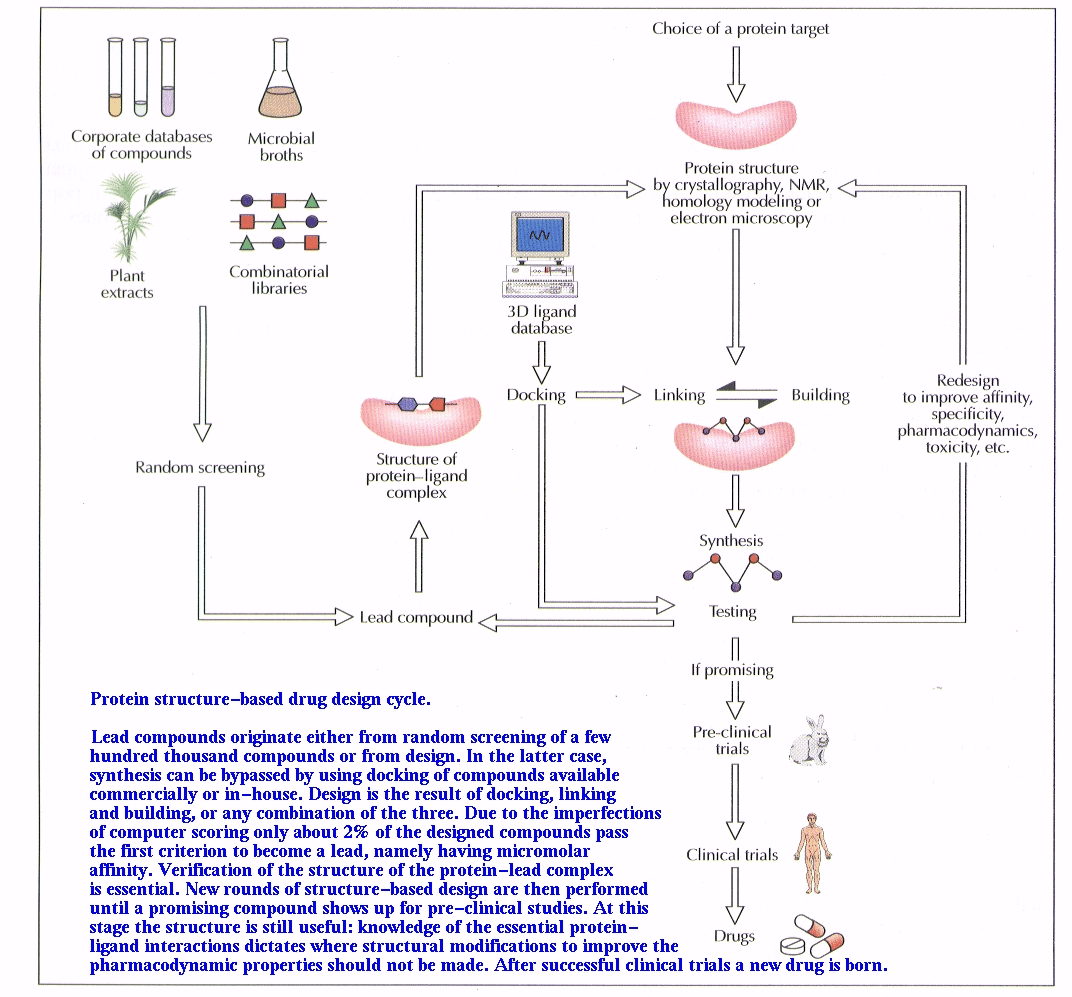
| Follow the links to see brief descriptions of research. Structure-based
Design of Drugs Structure-based
Design of Drugs |
Research in the Verlinde group is in the field of structure-based
drug design. In addition, Dr. Verlinde participates in the SGPP consortium,
a DNDI project, an MMV project, the Keck Center for Microbial Pathogens,
and performs 3D protein structure analysis studies for the Center for Ecogenetics
and Environmental Health (CEEH).
Group members:
Supported by NIH Grants GM-54,618 and AI-44,199. Group alumni:
|
{kind=link}